The first time I really tried to understand SARS-CoV-2 — not just follow the news, but actually understand it — I realized how much noise was drowning out the biology. Misinformation about treatments, confusing headlines about variants, vaccine debates with no mechanistic grounding. The virus itself was almost beside the point in most of what was being published.
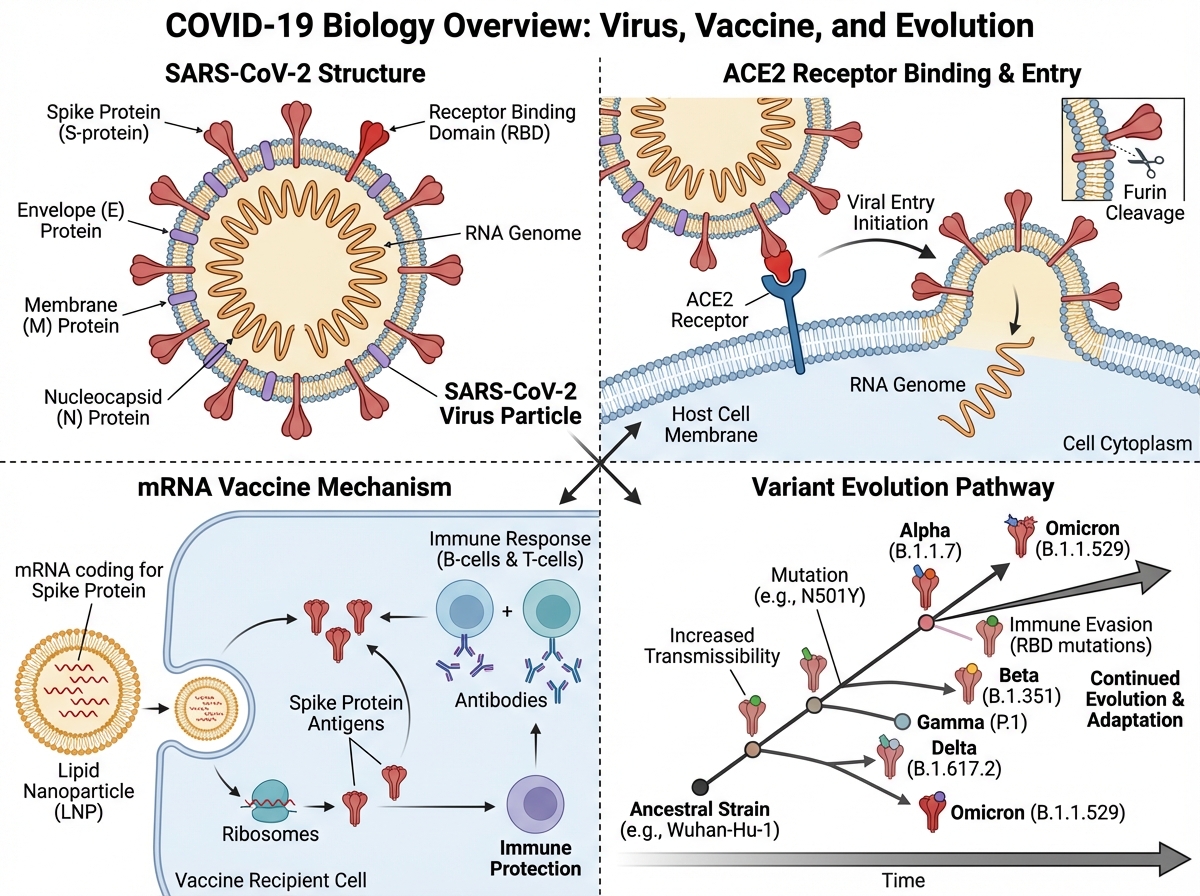
If you’re looking to learn how SARS-CoV-2 actually works — from its molecular structure to how vaccines were designed to stop it — the answer starts with the biology, not the politics. SARS-CoV-2 is a positive-sense single-stranded RNA betacoronavirus that enters human cells by binding its spike protein to the ACE2 receptor. Understanding that one mechanism unlocks almost everything else: why certain people got sicker, why variants emerged, why some treatments worked and others didn’t, and why mRNA vaccines could be developed so quickly.
- If you have a basic biology background, the molecular biology of SARS-CoV-2 is genuinely learnable — and it makes every future pandemic headline make sense faster.
- The real value isn’t memorizing facts about COVID-19 specifically — it’s building a framework for understanding any respiratory RNA virus.
- Most people who study this topic get stuck at treatments, skipping the immunology that makes vaccine logic click.
What Exactly Is a Virus — and Why Does It Matter Here?
A virus is not a cell. That distinction sounds obvious until you realize how many people expect antiviral drugs to work like antibiotics — and then get confused when they don’t. Viruses are obligate intracellular parasites: they carry genetic material (DNA or RNA), a protein coat called a capsid, and in some cases a lipid envelope, but they cannot replicate on their own. They need to hijack a host cell’s machinery to copy themselves.
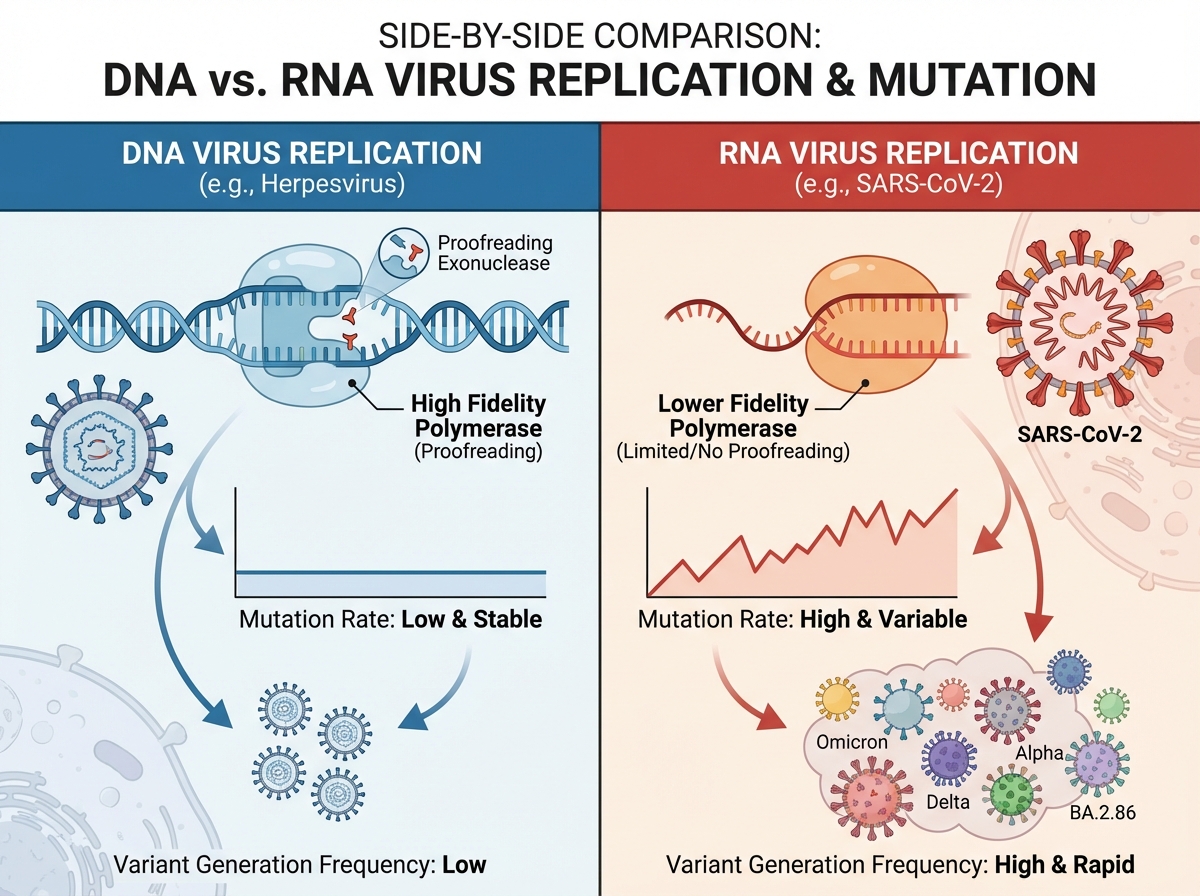
SARS-CoV-2 is an enveloped, positive-sense single-stranded RNA virus. “Positive-sense” means its RNA can be directly read by the host cell’s ribosomes as messenger RNA — no intermediate transcription step needed. This is part of why RNA viruses replicate so fast, and why they mutate so frequently: RNA polymerases lack the proofreading ability of DNA polymerases.
| Feature | DNA Viruses | RNA Viruses (like SARS-CoV-2) |
|---|---|---|
| Genetic material | Double-stranded DNA | Single-stranded RNA |
| Mutation rate | Lower | Higher |
| Replication speed | Slower | Faster |
| Examples | Herpesviruses, poxviruses | Influenza, HIV, coronaviruses |
Knowing this table isn’t trivia — it’s the foundation for understanding why SARS-CoV-2 generates variants, why flu vaccines need updating annually, and why HIV has proven so resistant to eradication.
Three Things About SARS-CoV-2 That Surprised Me
- The spike protein doesn’t just attach — it’s cleaved by the host’s own enzyme to activate entry.
- Variants aren’t random mutations; they’re evolutionary selections driven by immune pressure.
- The mRNA in COVID-19 vaccines degrades within days — it never reaches the nucleus.
How the Virus Actually Gets Inside You
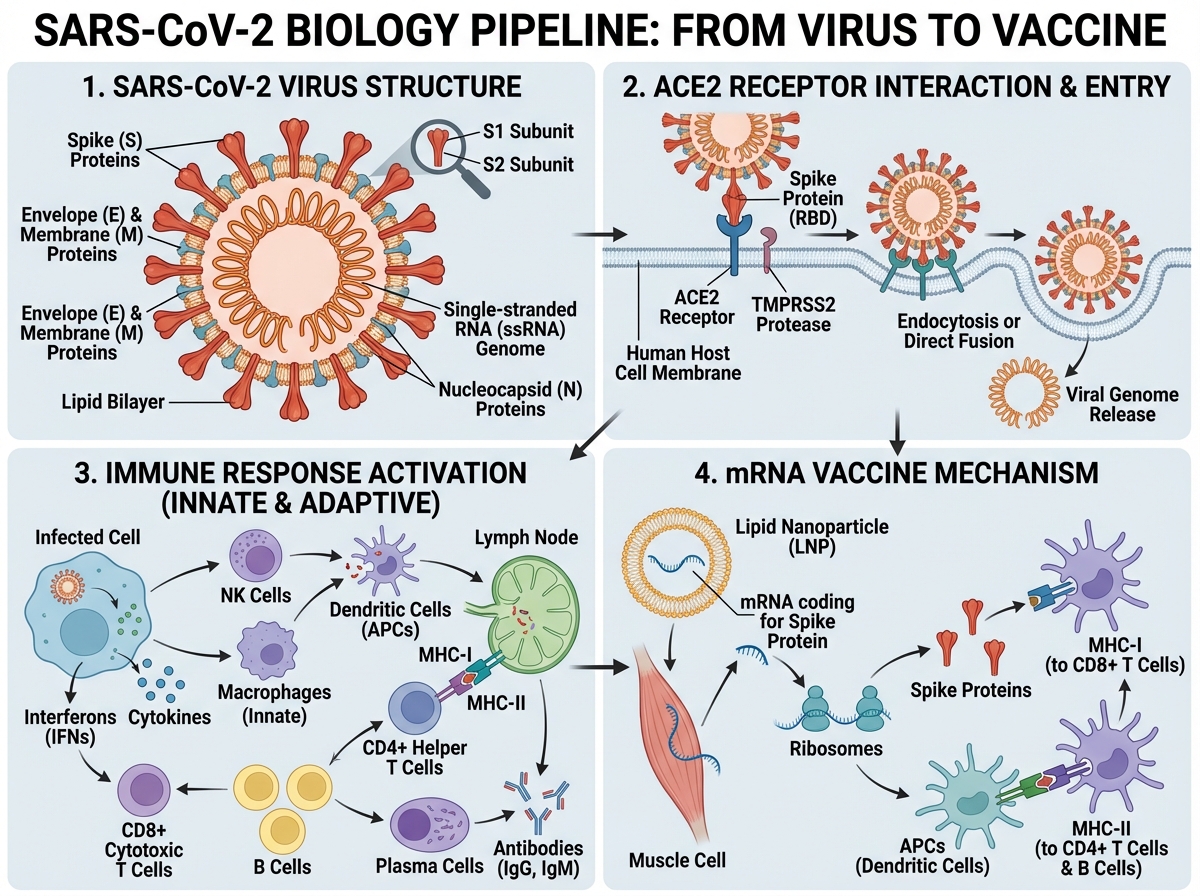
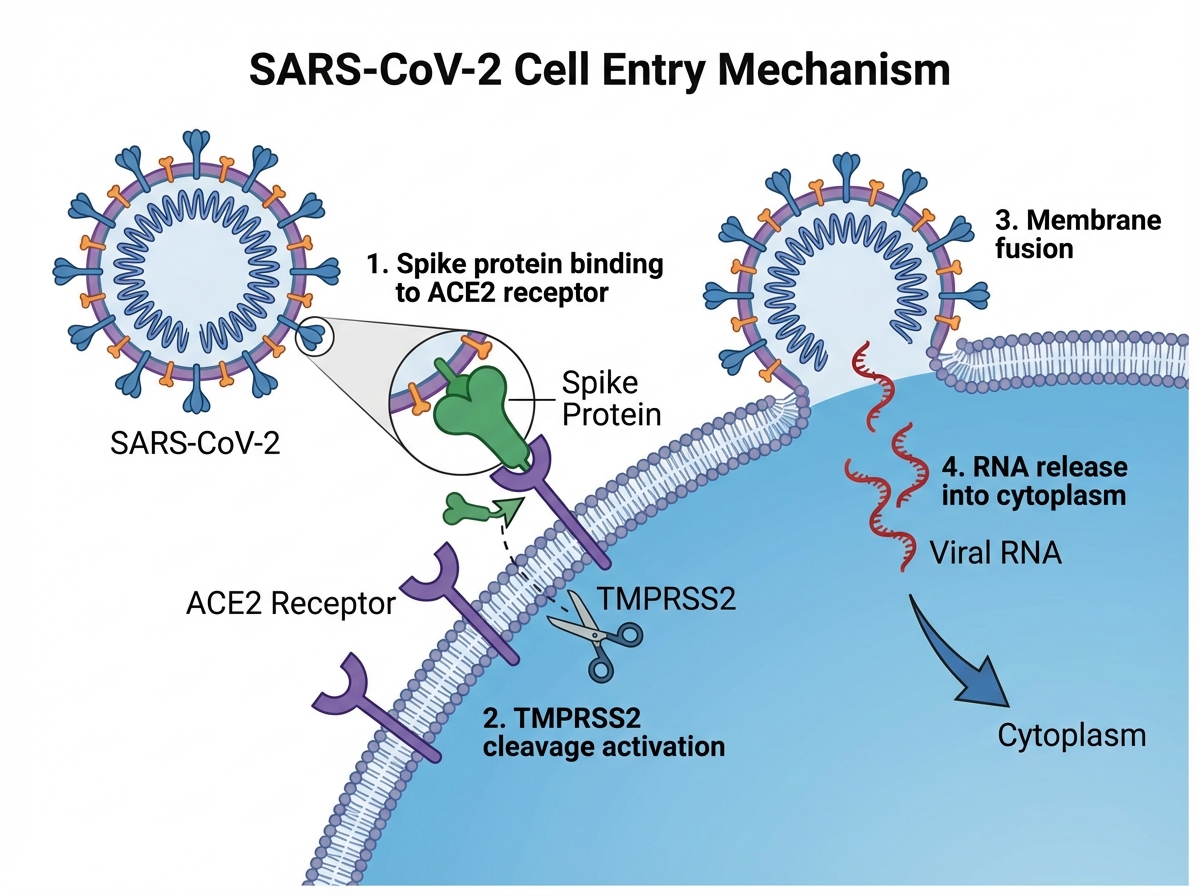
The spike protein is the key. SARS-CoV-2 uses it to bind ACE2 receptors, which are expressed heavily in the lungs, heart, kidneys, and gut — which explains the multi-organ involvement in severe COVID-19. But the binding step alone isn’t enough. A host enzyme called TMPRSS2 cleaves the spike protein at a specific site, triggering a conformational change that fuses the viral envelope with the cell membrane. The virus doesn’t break in — it gets let in by the cell’s own machinery.
Once inside, the viral RNA is released into the cytoplasm, where ribosomes immediately begin translating it. The virus encodes its own RNA-dependent RNA polymerase, which replicates the genome and produces subgenomic mRNAs encoding structural and accessory proteins. New virions assemble and bud from the cell, ready to infect neighboring cells — or be exhaled into the air.
This replication mechanism is clinically important. Drugs like remdesivir work by mimicking nucleosides, inserting themselves into the viral RNA strand and causing premature termination of replication. Knowing the mechanism tells you both why the drug has a narrow window of efficacy and why timing of administration matters.
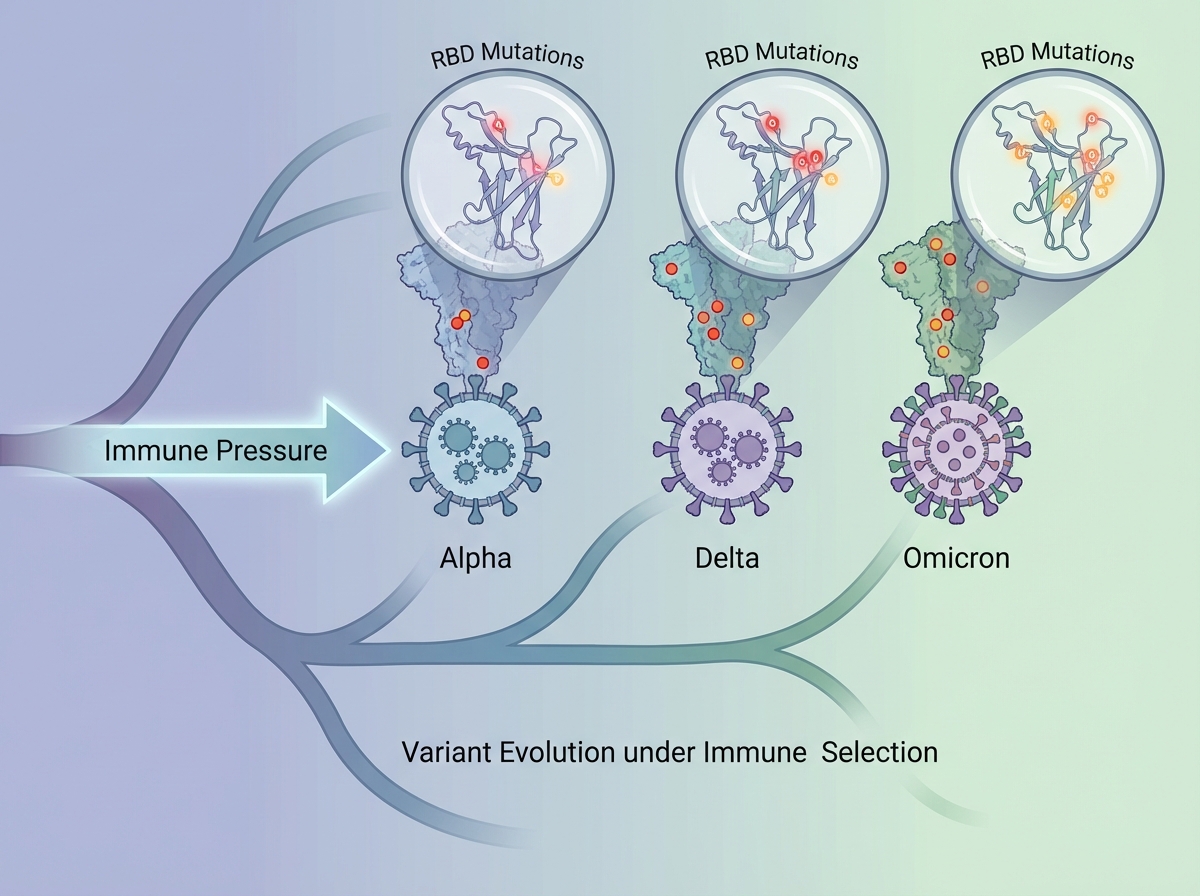
Why Variants Form — and Why That Was Always Going to Happen
The single biggest mistake people make when learning about viral variants is treating them as something unusual — a sign that SARS-CoV-2 was especially dangerous or unstable. Variants are normal. They’re inevitable for any RNA virus replicating at scale inside a large, partially immune population.
Every replication cycle introduces random mutations through the error-prone RNA polymerase. Most mutations are neutral or lethal to the virus. But some — particularly in the spike protein — improve the virus’s ability to bind ACE2, evade neutralizing antibodies, or replicate faster. In a population where increasing numbers of people have some immunity (from infection or vaccination), the variants that survive natural selection are precisely those with mutations that help them escape that immunity.
Alpha, Delta, Omicron — each wave represented a new dominant variant because those strains were better at spreading in the immune landscape of that moment. Omicron’s mutations were heavily concentrated in the spike protein’s receptor-binding domain, which is exactly where most vaccine-induced antibodies target. This is why booster doses and updated formulations became necessary, and it’s the same arms race that makes influenza vaccination an annual event rather than a one-time intervention.
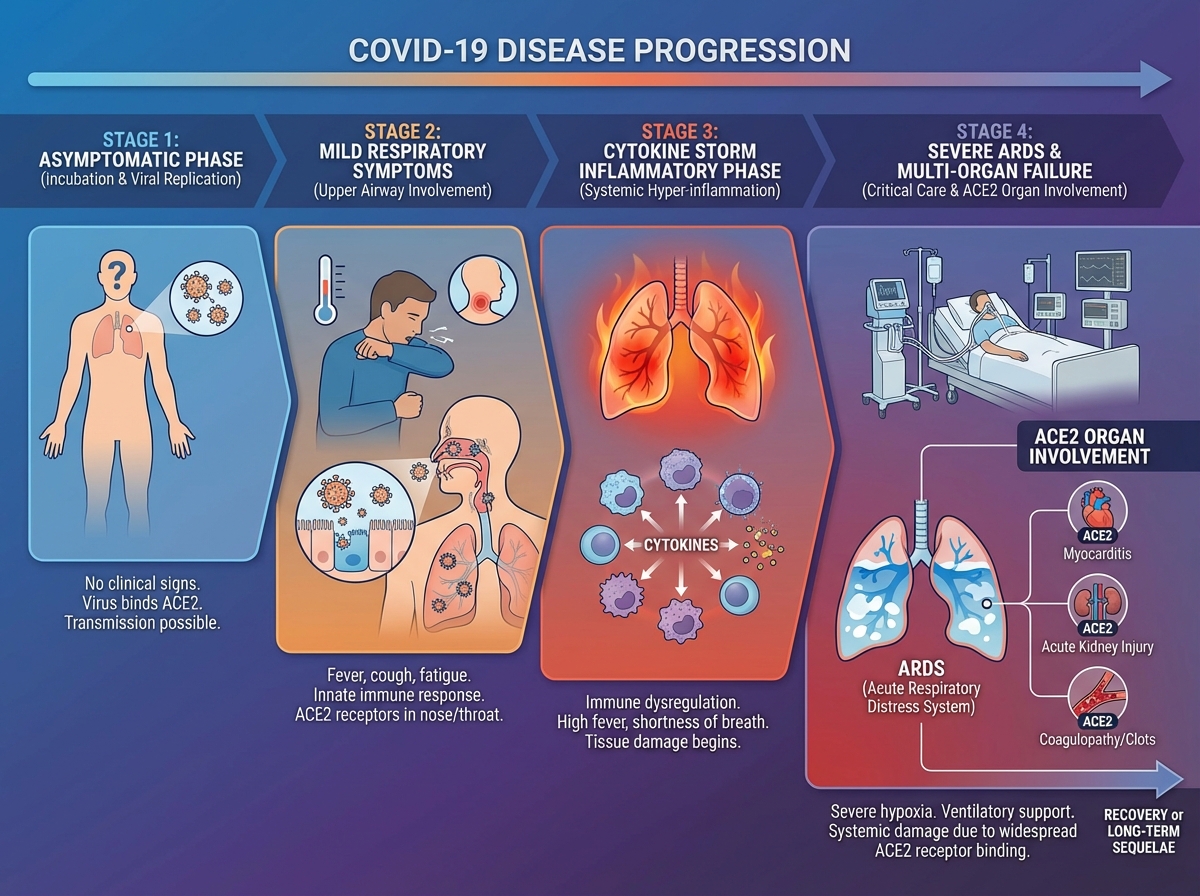
The Clinical Picture: Why Some People Got So Sick
COVID-19 is not simply a lung infection. The ACE2 receptor distribution explains why: damage isn’t confined to the respiratory tract. In severe cases, the disease progresses through a hyperinflammatory phase driven not purely by viral replication, but by the immune system’s own dysregulated response — a cytokine storm that causes damage disproportionate to viral load.
This is why dexamethasone, a corticosteroid, became one of the most evidence-backed COVID-19 treatments. It doesn’t target the virus directly — it dampens the inflammatory cascade that was killing patients on ventilators. The RECOVERY trial showed it reduced mortality in patients requiring oxygen or mechanical ventilation. Giving it too early, before the inflammatory phase, may actually blunt a necessary immune response. Treatment timing isn’t just a logistics question — it reflects the biology of disease stage.
Understanding the clinical spectrum — from asymptomatic to mild respiratory illness to severe pneumonia to multi-organ failure — requires understanding where the virus replicates, how the immune system responds, and at what point the immune response itself becomes the threat. That three-part framework explains most of the clinical variability observed across patients.
How to Learn SARS-CoV-2 Biology: The Progression That Actually Works
| Stage | Content | Time |
|---|---|---|
| Virology foundations | Virus types, replication basics, how pandemics spread | 1–2 hours |
| SARS-CoV-2 structure | Spike protein, ACE2 interaction, genome overview | 2–3 hours |
| Viral evolution and variants | Mutation mechanisms, selection pressure, variant tracking | 1–2 hours |
| Clinical and pathogenic aspects | Disease stages, cytokine storm, organ involvement | 2–3 hours |
| Treatment landscape | Drug development phases, repurposing, antivirals, antibody therapies | 2–3 hours |
| Vaccinology | Immunology principles, traditional vs. mRNA vs. viral vector approaches | 3–4 hours |
| Comparative virology | Influenza, HIV, pandemic risk framework | 1–2 hours |
| Total | Full foundational understanding | 12–19 hours |
The order here matters more than the pace. Jumping to vaccines without understanding the spike protein leaves you memorizing steps without understanding why each one works. Being slower than this estimate is completely normal — the treatment and vaccinology sections especially reward reading primary sources alongside structured learning.
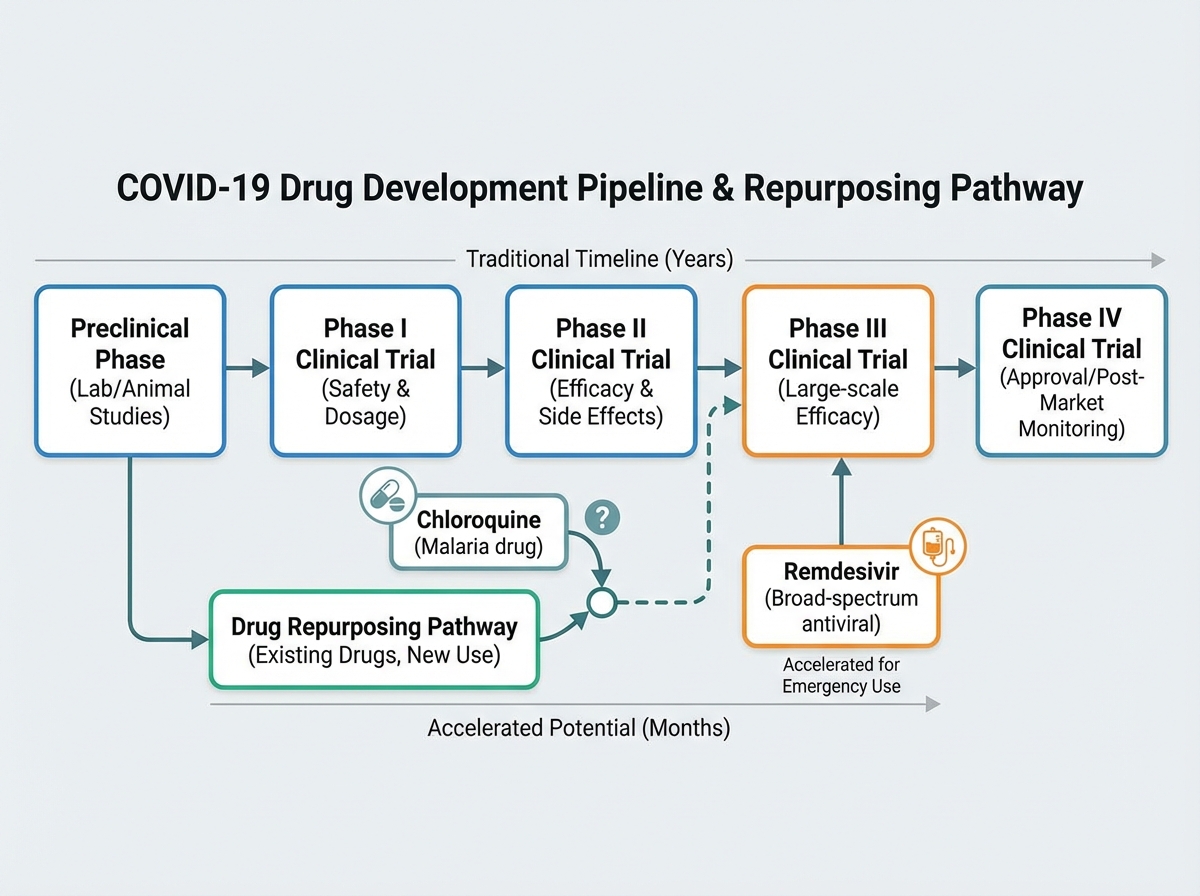
The Treatment Debate Was Never Really About the Drugs
Learning about COVID-19 treatments means confronting one of the messiest chapters in recent medical history. Chloroquine and ivermectin generated enormous controversy — not because the biology was ambiguous, but because the evidence pipeline was short-circuited by urgency, politics, and in vitro results that didn’t translate to clinical outcomes.
Chloroquine had a plausible mechanism: it raises endosomal pH, potentially blocking viral entry. In cell culture, it showed antiviral effects. But cell culture is not a human body. Phase III clinical trials — the ones designed to detect real-world efficacy and harm — didn’t support its use. The RECOVERY trial for hydroxychloroquine was stopped early due to lack of benefit. The lesson isn’t that the scientists who proposed it were dishonest; it’s that the drug development pipeline exists precisely because biological plausibility is not the same as clinical efficacy.
Antiviral drugs like remdesivir and later nirmatrelvir (Paxlovid) had more durable evidence. Monoclonal antibody therapies — engineered to neutralize the spike protein — worked well against earlier variants but lost effectiveness as Omicron’s spike mutations shifted the binding targets. Every treatment trajectory followed the same pattern: mechanism → preclinical → phased trials → real-world effectiveness. Understanding that pipeline makes you a more critical reader of any future treatment claim.
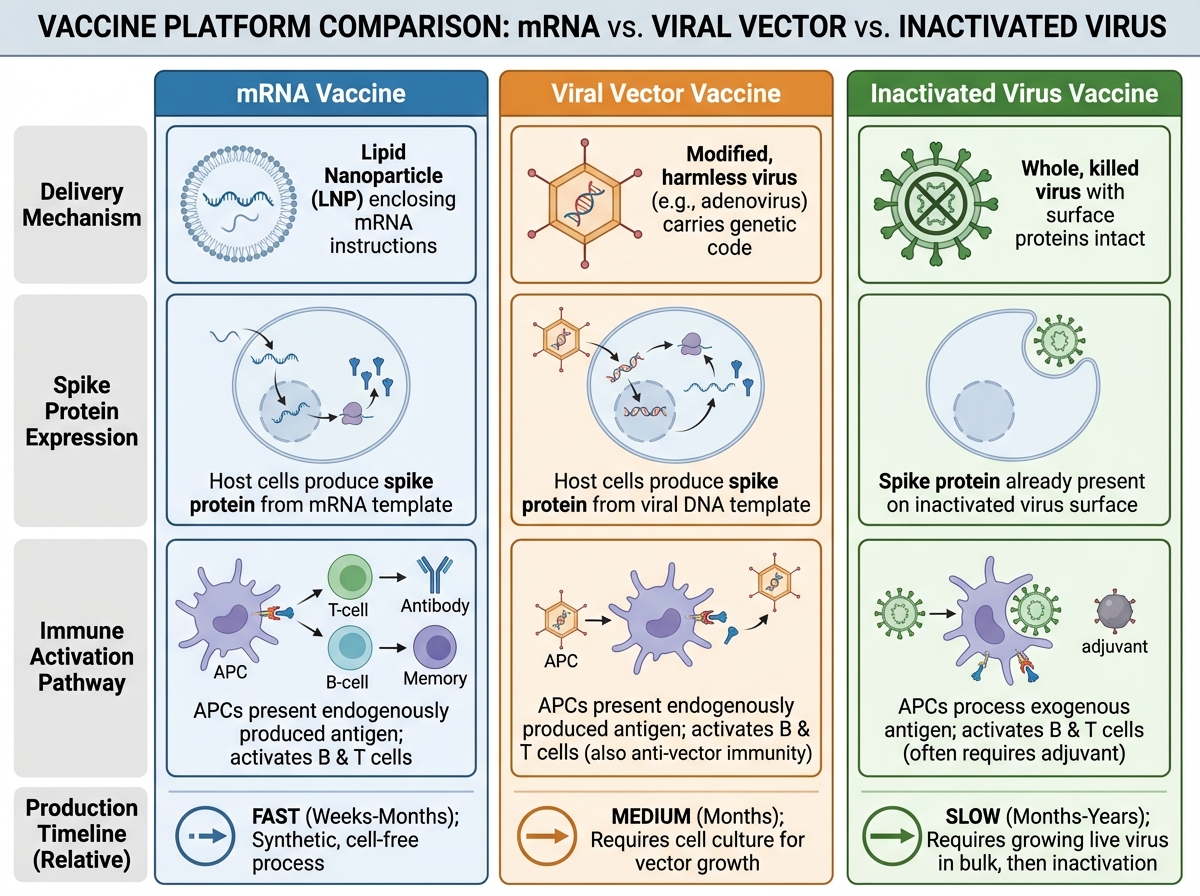
How mRNA Vaccines Actually Work — and Why the Speed Wasn’t Cutting Corners
The most common misunderstanding about mRNA vaccines is conflating speed with shortcuts. The development timeline was compressed not because safety steps were skipped, but because several phases ran in parallel (rather than sequentially) with massive funding, and because the mRNA platform itself had been in development for over a decade before COVID-19.
mRNA vaccines work by delivering instructions — not the virus itself. The mRNA sequence encodes the SARS-CoV-2 spike protein. Once injected, your cells translate that mRNA into spike protein, display it on their surface, and your immune system mounts a response: B cells generate antibodies, T cells develop memory. The mRNA itself is encapsulated in lipid nanoparticles to protect it from degradation, but it breaks down within days after delivery. It does not integrate into the genome — it never enters the nucleus.
Compared to traditional approaches — inactivated virus vaccines or protein subunit vaccines — mRNA platforms offer one major advantage: speed of design. Once you have the viral genome sequence, you can theoretically design an mRNA vaccine candidate in days. The manufacturing process is also more standardized than growing live virus. Viral vector vaccines (like AstraZeneca and Johnson & Johnson) use a modified adenovirus to deliver the same spike protein gene — a middle ground between traditional and mRNA approaches in terms of both mechanism and manufacturing complexity. For anyone interested in how options trading Greeks and implied volatility or financial modeling share a similar structure of layered complexity built on core mechanics, the logic of learning vaccine platforms bottom-up will feel familiar.
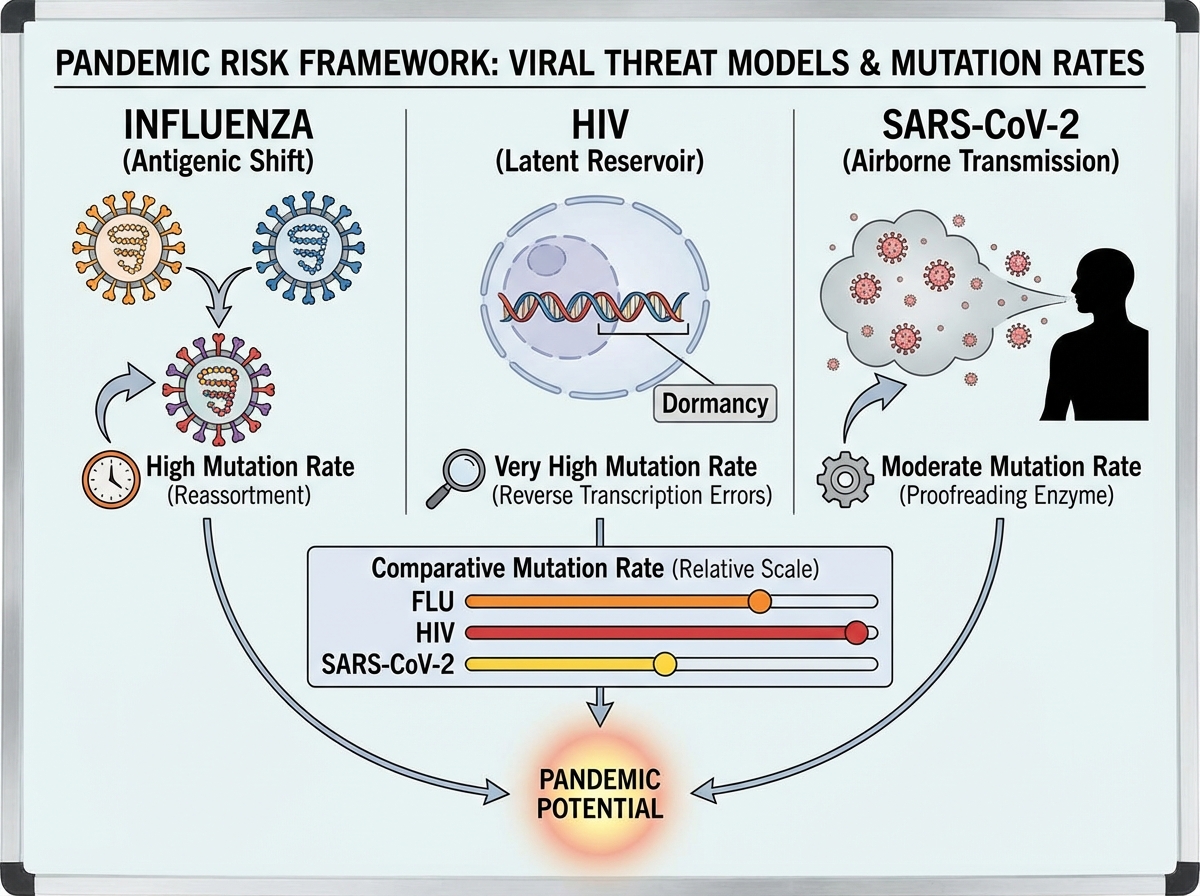
Beyond COVID-19: What Influenza and HIV Teach You About the Next Pandemic
Studying SARS-CoV-2 in isolation misses a bigger picture. Influenza and HIV each represent a different failure mode of pandemic control — and understanding both sharpens your framework for evaluating future threats.
Influenza undergoes both antigenic drift (gradual accumulation of point mutations) and antigenic shift (sudden reassortment of gene segments between strains). Antigenic shift is how the 1918 pandemic strain emerged, and it’s why a novel influenza with pandemic potential is taken seriously by epidemiologists every time a new avian or swine strain crosses into humans. The annual flu vaccine targets circulating strains, but a major antigenic shift could render it ineffective overnight.
HIV is a different kind of challenge: a retrovirus that integrates into the host genome, establishing a latent reservoir that no current therapy can fully clear. It mutates under antiviral pressure at an extraordinarily high rate — which is why HIV treatment requires combination antiretroviral therapy (cART) rather than single-agent drugs. Understanding HIV evolution gave virologists a template for thinking about escape mutations long before SARS-CoV-2 variants became a household concept.
Respiratory RNA viruses — coronaviruses and influenza especially — carry the highest pandemic risk because of their airborne transmission and rapid evolution. That combination, plus the density of human-animal interfaces in many regions of the world, means the question is not whether another pandemic will emerge, but when and which pathogen.
What Actually Matters When You Look Back on This
Learning the biology of SARS-CoV-2 changed how I read every health headline. Not because I became an expert, but because I understood the mechanism well enough to ask the right question: what phase is this evidence from? What does the proposed drug actually target? Why would this variant behave differently from the last?
That shift from passive consumption to mechanistic thinking is the real outcome. The specific facts about COVID-19 will continue to evolve — new variants, updated treatments, revised recommendations. But the biological framework — how RNA viruses replicate, how immune memory forms, how drug development phases work — remains stable. That’s what’s worth carrying forward.
For biomedical and bioscience students specifically, understanding SARS-CoV-2 end-to-end is not just a pandemic postmortem. It’s a case study in applied virology, pharmacology, immunology, and epidemiology compressed into a single, well-documented event. That case study doesn’t end — it continues every time a new variant emerges or a new antiviral enters trials.
Start with the spike protein, not the symptoms. Understanding ACE2 binding before clinical presentation gives you a causal model instead of a symptom checklist.
Use NCBI to look at the actual viral genome. Reading a real SARS-CoV-2 RNA sequence in a public database makes mutation discussion concrete instead of abstract.
Learn the four phases of drug development before evaluating any treatment claim. Phase I data and Phase III data are not the same thing — and most COVID-19 treatment controversies live in that gap.
Separate the inflammatory phase from the replication phase clinically. Antivirals are most effective early; immunosuppressants like dexamethasone are most effective late. Mixing them up is a conceptual error with real consequences.
Trace each vaccine type back to its delivery mechanism. mRNA, viral vector, and inactivated virus vaccines all target the same protein but take completely different routes — knowing the route explains the differences in storage, dosing, and variant sensitivity.
Track epidemiological data from primary sources. WHO, CDC, and Our World in Data give you raw numbers; learning to read them directly removes the interpretive layer that most media adds.
When a new variant emerges, check its spike protein mutations first. Specifically the receptor-binding domain — that’s where most immune escape happens and where most neutralizing antibodies bind.
Connect influenza and HIV to SARS-CoV-2 as a comparative framework. Each virus represents a different evolutionary strategy, and holding all three in mind sharpens your instinct for which new pathogens deserve serious pandemic concern. If your broader interests include learning how complex systems are modeled and predicted — whether biological or financial — the approach behind stock market prediction using cycle-based systems uses similar pattern-recognition logic applied to a different domain.
Leave a Reply