There’s a particular kind of discomfort that comes from sitting across from a patient whose family history is a tangle of early-onset cancers and not knowing what to do with that information. Not because you’re unaware that genetics matters — you know it does — but because the distance between knowing that and acting on it clinically is wider than any textbook suggests.
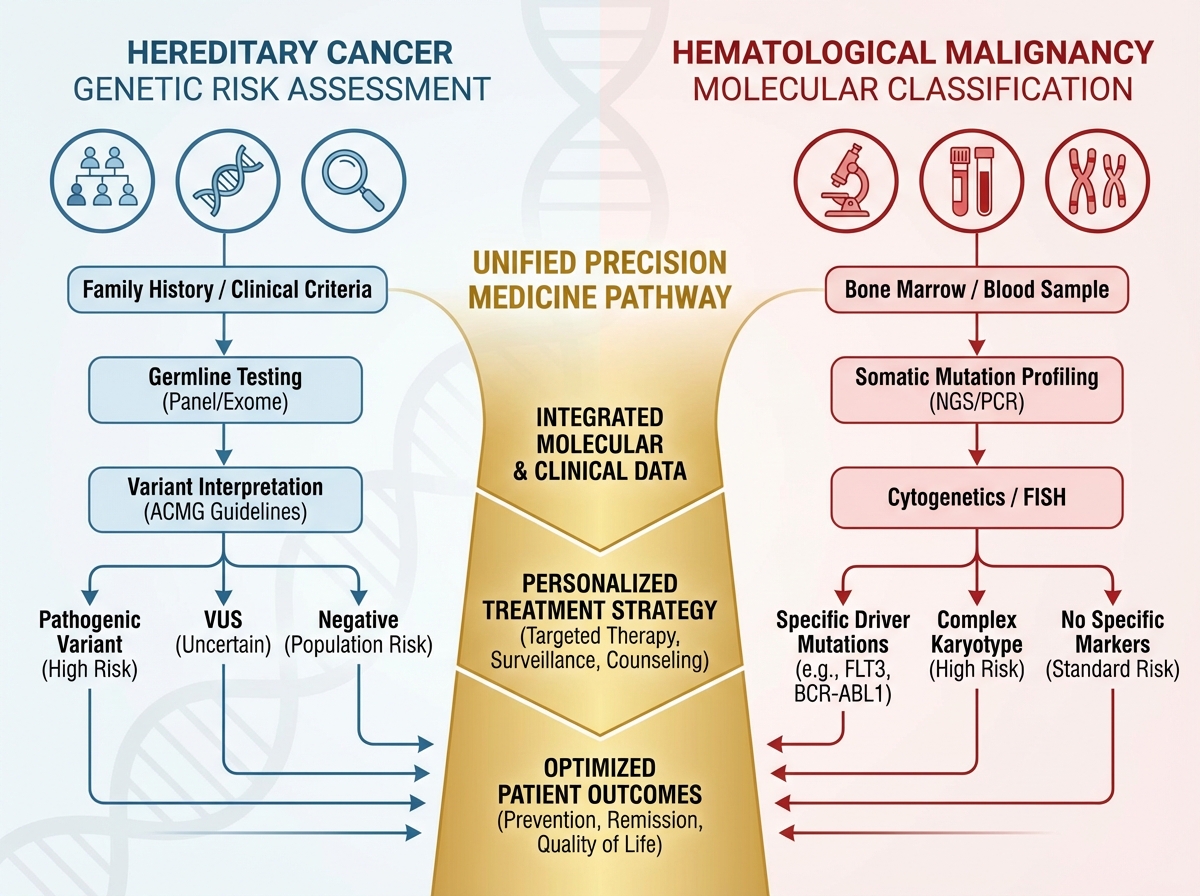
If you’re looking to understand hereditary cancers and hematological malignancies at a clinical and molecular level, the starting point is accepting that these two fields — once treated as separate silos — are converging around the same core challenge: identifying who is at risk before disease declares itself, and then acting with precision when it does. Hereditary cancer genetics gives you the upstream framework; hematological oncology forces you to apply molecular thinking in real time, often under diagnostic pressure. Together, they represent the current frontier of precision medicine in oncology.
- Knowing a patient carries a susceptibility allele changes nothing unless your institution has a clear testing and referral pathway in place.
- The biggest gap in hematological cancer care isn’t treatment — it’s the delay between symptom onset and molecular diagnosis.
- Precision medicine in hematology only works when the diagnostic data is granular enough to actually differentiate subtypes.
What “Hereditary Cancer” Actually Means at the Genetic Level
The term gets used loosely, and that looseness causes real clinical problems. A hereditary cancer predisposition isn’t simply “cancer that runs in the family.” It refers specifically to an inherited germline variant — typically in a tumor suppressor gene or DNA repair gene — that significantly elevates lifetime cancer risk compared to the general population.
These variants are stratified by penetrance:
| Penetrance Type | Risk Elevation | Examples |
|---|---|---|
| High penetrance | >50% lifetime risk | BRCA1/2, MLH1, APC |
| Intermediate penetrance | 20–50% lifetime risk | CHEK2, ATM, PALB2 |
| Low/susceptibility alleles | Modest population-level risk | Common SNPs in GWAS studies |
For the clinician or researcher entering this space, the penetrance distinction isn’t academic — it directly determines whether a variant finding triggers prophylactic intervention, enhanced surveillance, or simply a note in the chart. Conflating high-penetrance variants with susceptibility alleles is one of the most common and consequential errors in genetic counseling practice.

Three things that are less obvious than they should be:
- A negative genetic test never rules out hereditary risk — it only rules out tested variants.
- Most hereditary cancer syndromes are autosomal dominant, but penetrance is modified by environment and other genetic factors.
- Panel testing has outpaced the clinical infrastructure needed to interpret its results.
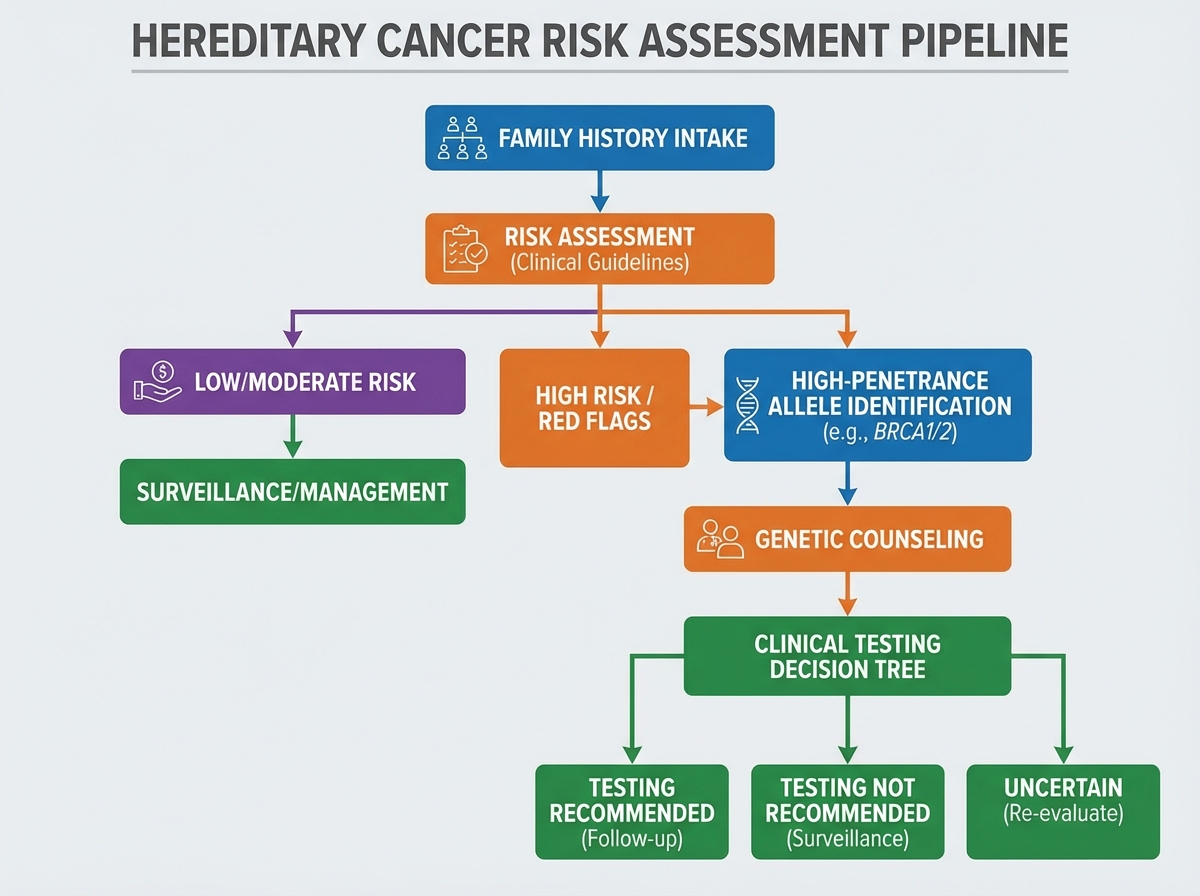
The Learning Curve on Genetic Testing Criteria
The first time you work through the diagnostic criteria for hereditary cancer testing, they feel logical. Then you apply them to an actual patient and realize the criteria were designed for ideal family histories — complete, accurate, and multi-generational. Most family histories are none of those things.
The standard frameworks — Amsterdam criteria for Lynch syndrome, NCCN guidelines for BRCA testing — give you a starting structure. But what they don’t prepare you for is the ambiguity that dominates real-world assessment: the patient whose maternal relatives all died young from unrelated causes before cancer could manifest, the family with no documentation across generations, the patient adopted at birth. These aren’t edge cases. They’re routine.
The realization that finally changes how you approach this is understanding that genetic testing criteria are risk-stratification tools, not gatekeeping checklists. The question isn’t “does this patient meet criteria” — it’s “what is this patient’s pre-test probability, and does testing change management?” Once you reframe the decision that way, the criteria become one input among several rather than a pass/fail threshold.
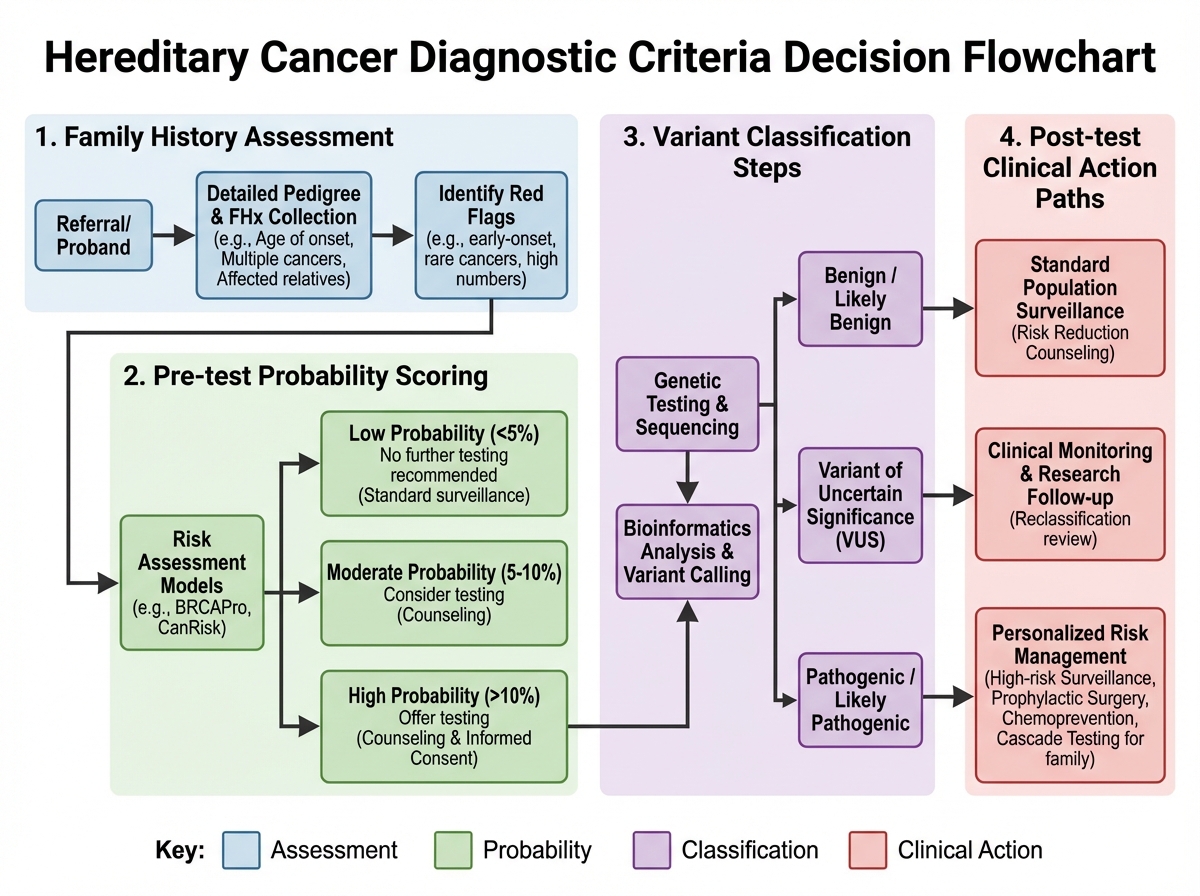
This shift also changes how you read a variant of uncertain significance (VUS). A VUS on a panel report is not a finding — it’s an open question. Treating it as actionable is one of the most common mistakes made by clinicians who are new to germline testing interpretation, and it generates real patient harm through unnecessary surgeries and anxiety.
Where Hereditary Cancer Testing Still Falls Short
Here’s where the field gets uncomfortable: the infrastructure for hereditary cancer testing has not kept pace with the biology. Genetic testing technology advanced faster than the clinical frameworks for deploying it, interpreting it, and acting on it equitably.
The knowledge gaps are real and specific. Many variants — particularly in genes like PALB2 or RAD51C — still lack sufficient evidence to assign clear clinical action thresholds. The databases that classify variants are built primarily from populations of European ancestry, which means pathogenicity classifications for the same variant can differ meaningfully across ethnic groups. This isn’t a minor technical limitation — it’s a structural equity problem embedded in the evidence base.
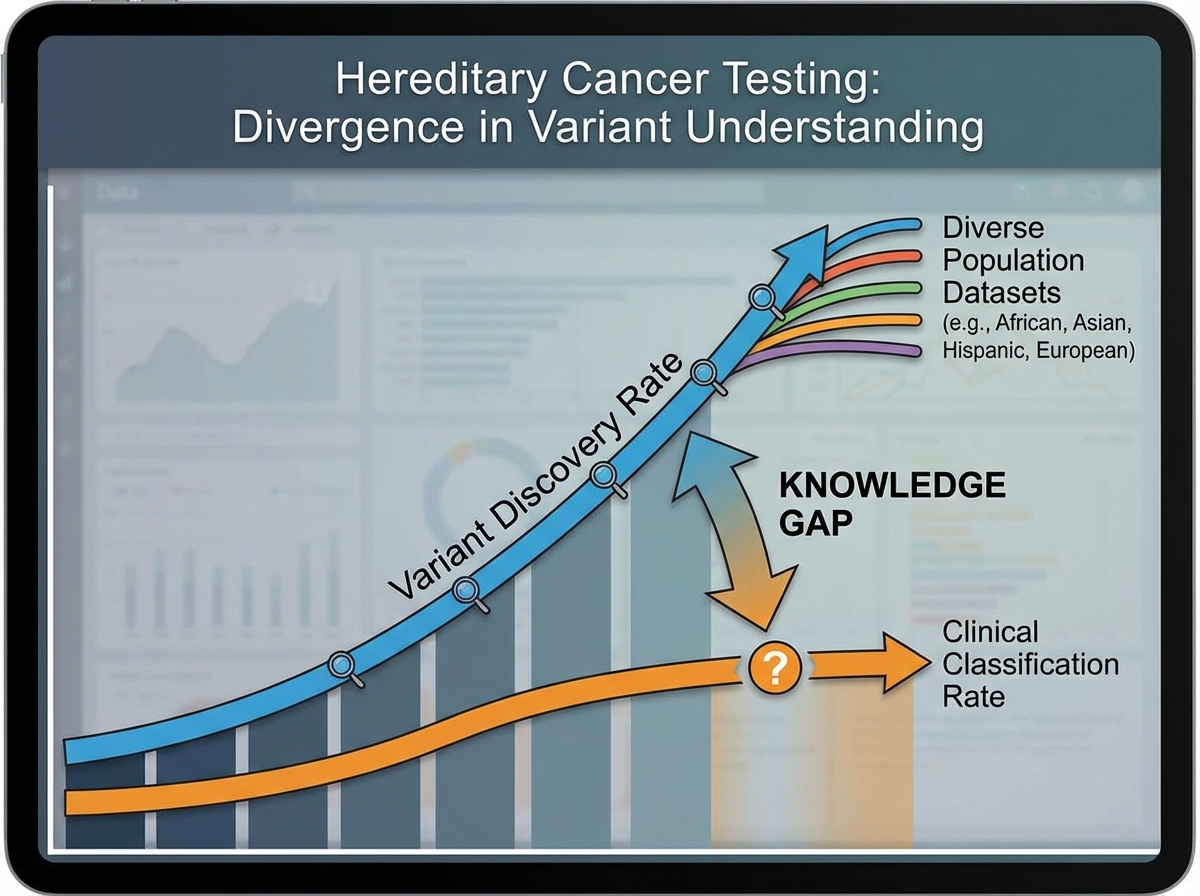
Barriers to testing access compound the problem. Genetic counselors are unevenly distributed geographically. Reimbursement for panel testing varies by payer and indication. Patients who would benefit most from cascade testing — relatives of known variant carriers — are often never contacted. The future direction here isn’t more testing; it’s smarter deployment of testing within systems that can actually support the downstream care.
How Hematological Cancers Demand a Different Kind of Diagnostic Thinking
Moving from hereditary cancer genetics into hematological malignancies feels like shifting registers. The hereditary side is fundamentally about probability and prevention. Hematological oncology is about classification under pressure — you often have a very sick patient, a complex morphology report, and a treatment decision waiting on a molecular result.
The epidemiology of hematological cancers already tells you something important about their complexity: they encompass leukemias, lymphomas, and myelomas — each a family of diseases, not a single entity. Acute myeloid leukemia (AML) alone has over a dozen molecularly defined subtypes with different prognostic profiles and therapeutic targets. Treating AML as a single disease is the equivalent of treating all carcinomas the same way.
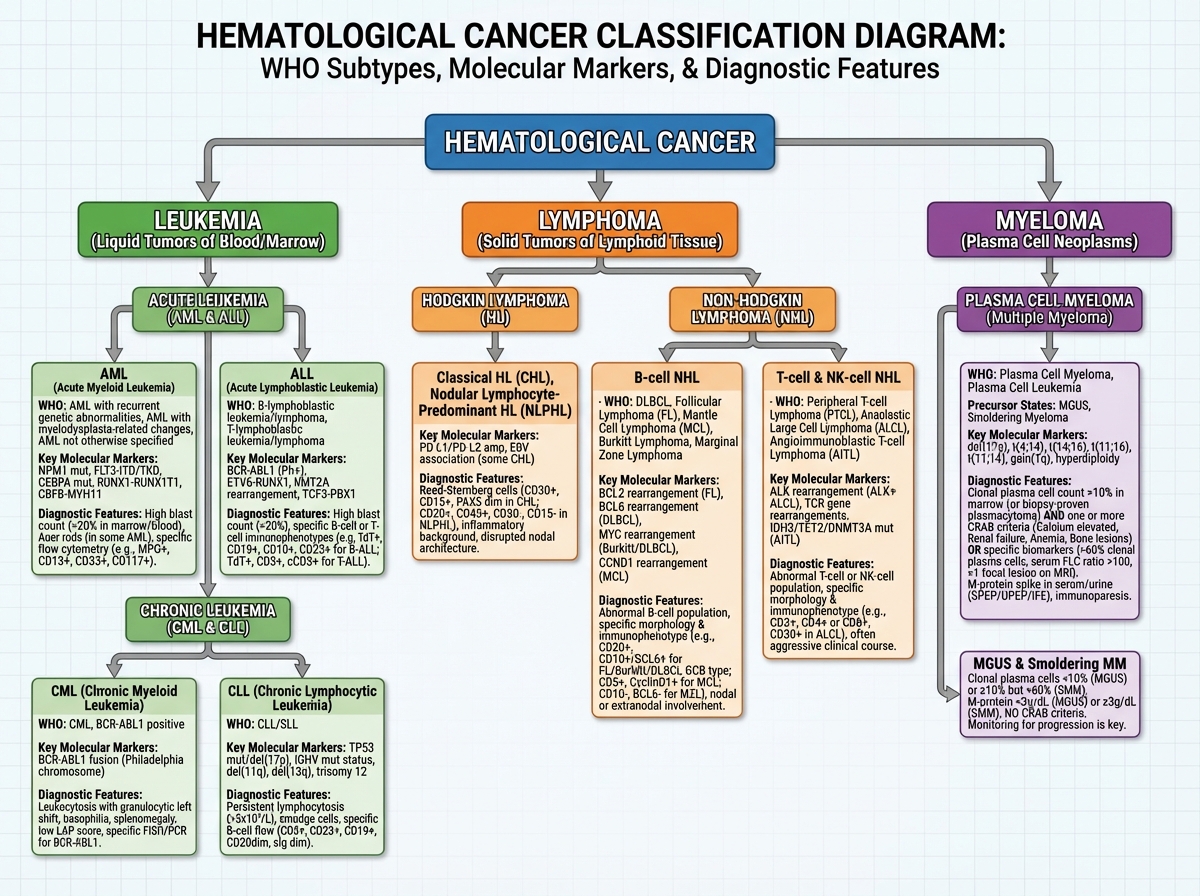
What changes when you understand this at a molecular level is how you read a diagnostic workup. Morphology is the entry point, not the conclusion. Flow cytometry, cytogenetics, FISH, and next-generation sequencing aren’t confirmatory — they’re definitional. The diagnosis isn’t complete until the molecular characterization is complete, because the molecular profile is what determines treatment eligibility, prognosis, and monitoring strategy.
The Role of Molecular Characterization in Hematological Diagnosis
The biggest mistake people make when entering hematological oncology is assuming that a diagnosis is established once a malignancy is confirmed. That framing made sense twenty years ago. Today, confirming the presence of leukemia without characterizing it molecularly is roughly equivalent to confirming a patient has an infection without identifying the organism.
Molecular characterization in hematological cancers serves three distinct clinical functions: prognostic stratification, therapeutic targeting, and minimal residual disease (MRD) monitoring. These functions are not interchangeable, and not every marker serves all three. FLT3-ITD in AML is both a prognostic marker and a therapeutic target — FLT3 inhibitors are now standard of care. NPM1 mutation, on the other hand, is primarily prognostic and serves as a sensitive MRD marker due to its stability across disease course.
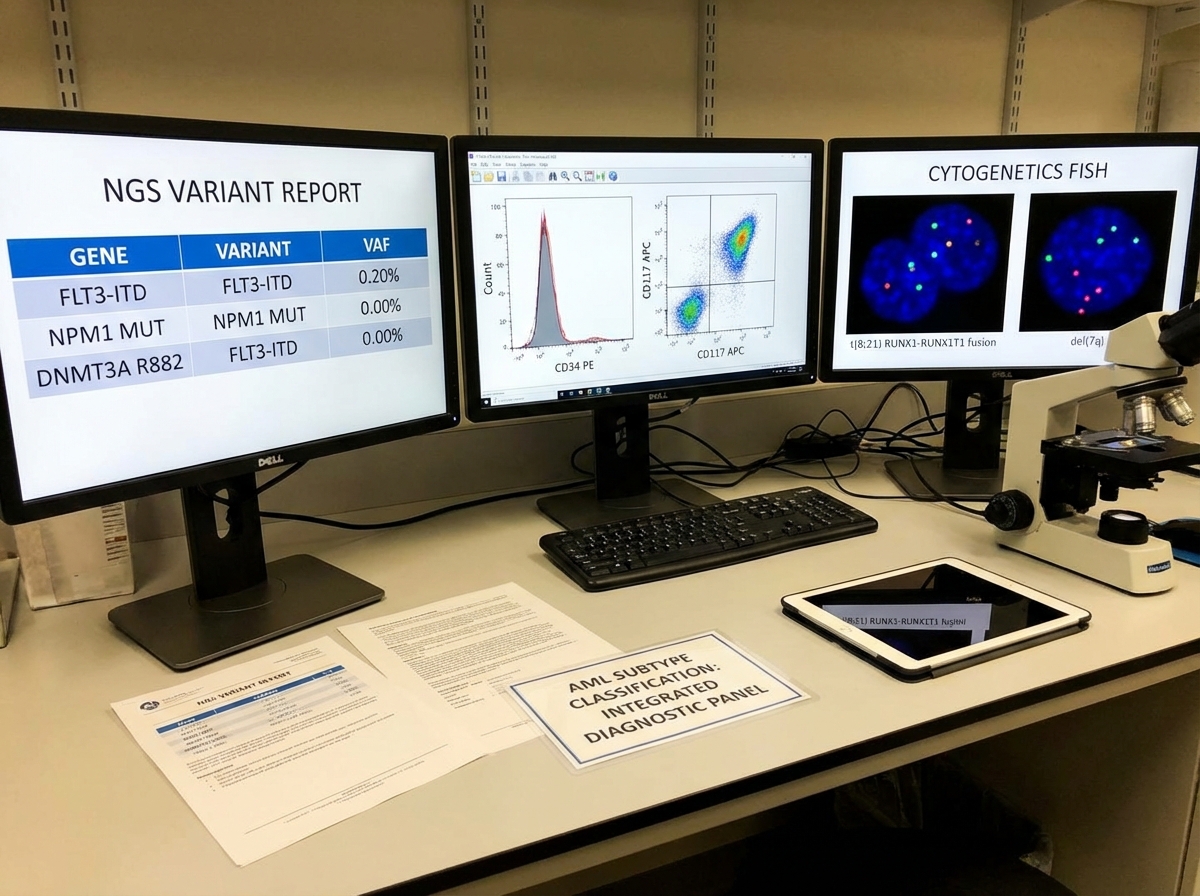
T-cell acute lymphoblastic leukemia (T-ALL) illustrates how granular this thinking has to be. T-ALL is biologically and clinically distinct from B-ALL despite sharing the same disease category label. Its molecular subtypes — driven by different transcription factor rearrangements and signaling pathway activations — have different relapse patterns and different sensitivities to specific agents. MRD assessment in T-ALL requires assay sensitivity calibrated to the specific clonal marker, which is why the choice of MRD methodology matters as much as the decision to monitor at all.
Precision Hematology: What It Requires Beyond the Science
The phrase “precision medicine” is used so freely in oncology that it has started to lose operational meaning. In hematology, precision medicine has a specific requirement that often gets glossed over: it only works when the diagnostic data is granular enough to actually differentiate subtypes, and when the clinical infrastructure exists to act on that differentiation.
Functional precision medicine takes this further. Rather than relying solely on genomic profiling to predict drug sensitivity, functional approaches test patient-derived cancer cells ex vivo against a panel of agents to directly measure response. This is particularly valuable in hematological malignancies where tumor heterogeneity and clonal evolution make genomic predictions unreliable over time.
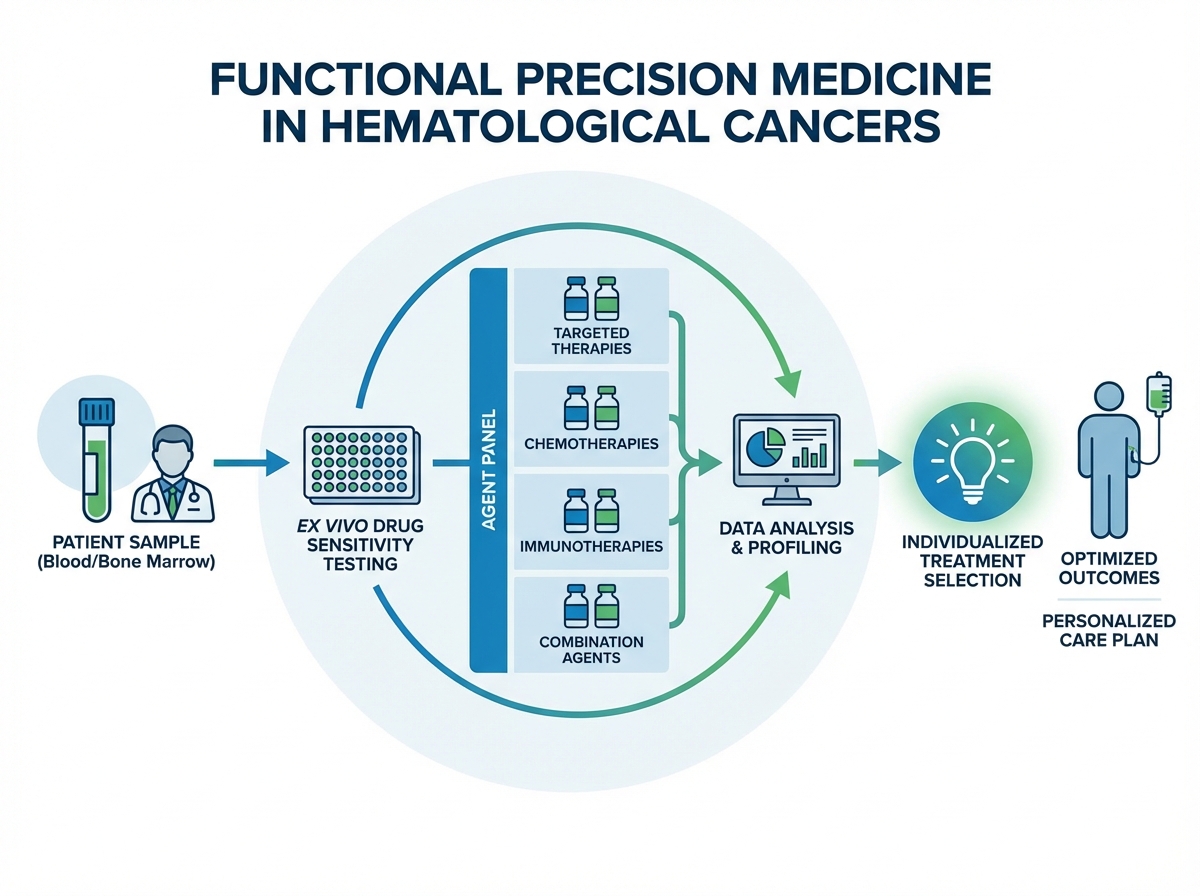
The practical challenge is implementation. Functional assays require living cells, which means sample logistics matter. Turnaround time has to align with clinical decision windows. Standardization across institutions is incomplete. These aren’t reasons to dismiss the approach — they’re the specific problems that need solving for precision hematology to deliver on its promise at scale. Understanding them clearly is what separates a clinician who can use these tools from one who simply knows they exist.
Diagnosis Delays and the Healthcare System Cost No One Talks About Enough
There is a policy dimension to both hereditary cancers and hematological malignancies that practitioners often defer to administrators, and that’s a mistake. The delay between symptom onset and accurate molecular diagnosis in hematological cancers is not just a clinical problem — it is a measurable driver of healthcare cost, worse outcomes, and treatment complexity.
Patients who receive a precise molecular diagnosis early enter treatment at an earlier disease stage, with clearer therapeutic options and lower rates of treatment-related complications from empirical or mismatched therapies. The inverse is also true: delayed or imprecise diagnosis means more advanced disease at treatment initiation, higher rates of relapse, and longer, more resource-intensive treatment courses.
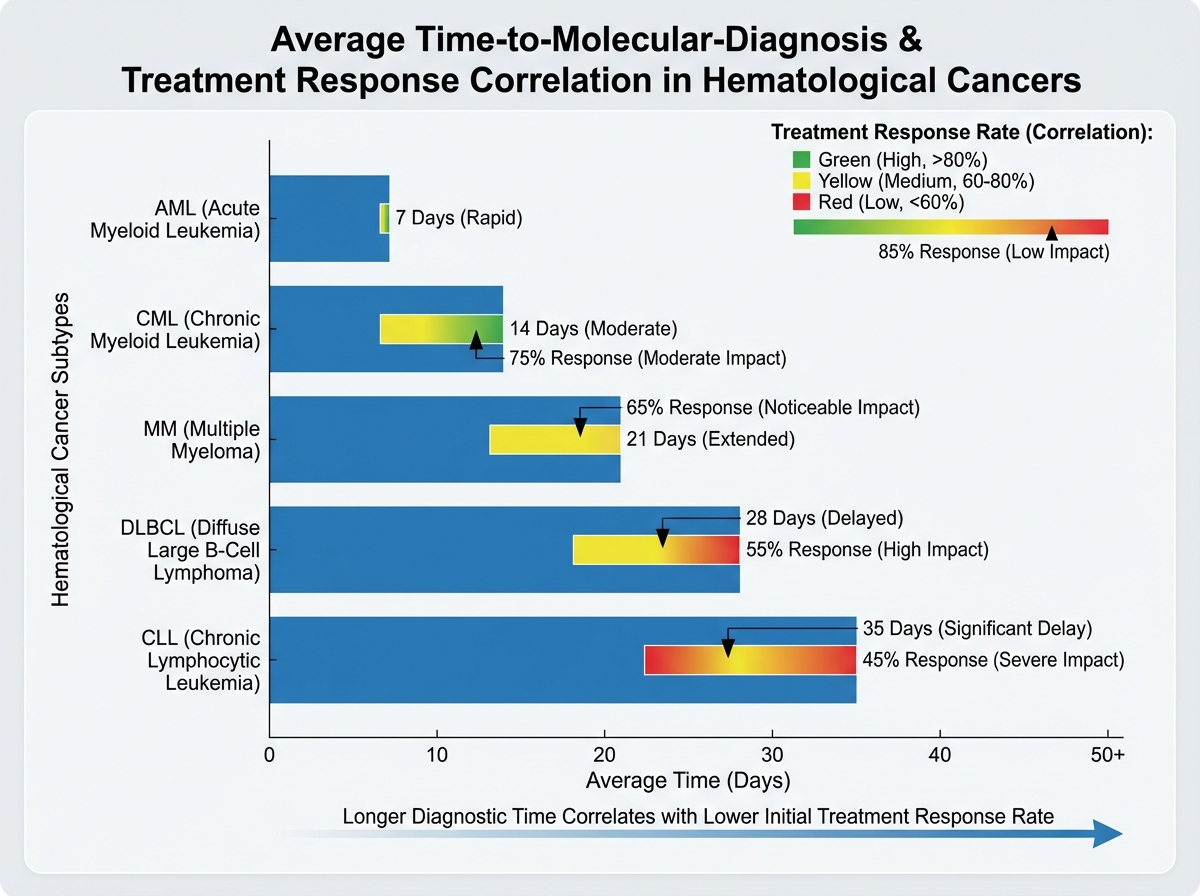
For hereditary cancers, the system-level cost of under-testing is even clearer. Identifying a BRCA1 carrier before cancer develops costs far less — in both human and economic terms — than treating the metastatic breast or ovarian cancer that follows from missing the risk. Cascade testing of family members after a proband is identified is one of the highest-value interventions in preventive oncology, yet uptake remains low in most health systems. The gap between what the science supports and what the system delivers is not a scientific problem — it’s a structural and policy one.
Looking back at how much of this field sits at the intersection of molecular biology, clinical decision-making, and health system design, what stands out is how much the practitioner’s role has expanded. You’re no longer just applying knowledge — you’re navigating between incomplete evidence, real patients, and systems that haven’t fully caught up with the science.
Here’s what actually helps when you’re working in this space:
- Map your institution’s germline testing pathway before you need it. Knowing who orders, who counsels, and who acts on results in advance prevents the delay that happens when you’re trying to figure it out in front of a patient.
- Learn to read a variant classification report, not just the summary. The difference between pathogenic, likely pathogenic, and VUS has direct clinical consequences — don’t let that interpretation happen upstream without your input.
- Treat molecular characterization as part of the diagnosis, not a follow-up. In hematological malignancies, the molecular profile is the diagnosis. Build your workflow so it’s concurrent, not sequential.
- Understand MRD methodology before ordering MRD testing. Different assays have different sensitivity thresholds and different clinical validation levels. The result only means something if the method is appropriate for the disease context.
- Know the penetrance tier of any variant you’re acting on. High-penetrance and intermediate-penetrance variants require different clinical responses — conflating them creates both under- and over-treatment.
- Build family history into the consultation, not the chart review. A family history taken by a clinician who knows what they’re looking for yields different information than one documented by an intake form.
- Follow functional precision medicine trial data actively. This is moving fast. What’s investigational today in hematological oncology has a track record of becoming standard of care within a single training cycle.
- Push for cascade testing protocols in your institution. Identifying a variant in a proband without a system to reach their relatives captures only a fraction of the preventive value — make that the standard, not the exception.
Leave a Reply